
MOLECULAR DESIGN AND CLONING OF SCRAMBLED SMALL-HAIRPIN RNA FOR KNOCKING DOWN HUMAN GELATINASE B
Mogulevtseva J.A., Bachelor of Science, Federal Russian State Agrarian University — Moscow Timiryazev Agricultural Academy (Moscow, Russia), student, bioengineering.
Mezentsev A.V., Doctor of Philosophy N.I. Vavilov Russian Institute of General Genetics – IOGen RAS (Moscow, Russia), senior scientist, medical genetics.
ABSTRACT
Matrix metalloproteinases are a group of proteolytic enzymes that are important for the maintenance of skin homeostasis. The aim of this paper was to design and clone the sequence encoding scrambled small-hairpin RNA (shRNA) that can be used as a negative control in silencing the gene of human gelatinase B. Alternative sequences of shRNA were compared using the online tools «Blastn», «Oligo Calc» and «Palindromic sequences finder». Double stranded DNA encoding the selected shRNA was obtained by annealing of two complementary single stranded oligonucleotides. Commercial T4 DNA ligase and restriction endonucleases BamH1 and EcoRI were used to clone the annealed oligonucleotide into the expression vector pGPV-17019250. Thus, we designed scrambled shRNA that can be used as a counterpart of specific shRNA in silencing the gene of human gelatinase B and obtained the expression vector pGPV-17019250-KGB for its expression in cultured human cells.
Key words: human gelatinase B, cloning, vector, scrambled shRNA, psoriasis.
Introduction
Matrix metalloproteinases (MMPs) are a group of zinc-containing, calcium-dependent endopeptidases [6]. Their physiological role is unique due to their broad substrate specificity. Particularly, MMPs are capable of processing the proteins of extracellular matrix (ECM), activating growth factors and proenzymes, as well as the specific receptors located on the cellular membrane.
In psoriasis, MMPs contribute to remodeling of skin and dermal microcapillaries [6]. MMPs secretion by immune cells let them penetrate the blood vessels and invade the inflamed skin. Moreover, high expression of gelatinase B (MMP9) in lesional skin of psoriasis patients correlates with disease severity [8]. In this respect, controlling MMPs expression in diseased cells may have a clinical value and be beneficial for psoriasis patients. Presumably, at least two standard approaches could be used to control MMPs activity. Firstly, MMPs could be inhibited by specific inhibitors. Secondly, MMPs biosynthesis on ribosomes could be suppressed by small interfering RNAs (siRNAs) capable of destroying the MMPs-specific mRNAs. The aim of this study was to obtain a vector that would express scrambled shRNA. In perspective, this scrambled shRNA could be used as a counterpart of the specific shRNA directed to human gelatinase B [4].
Materials and methods
To design of shRNA, the sequence of previously designed GB siRNA [4] was transformed into two different sequences of scrambled siRNAs using «siRNA Wizard Software» (InvivoGen, USA) and «Sequence Scramble» (GenScript, USA) online tools. «Blastn» (NCBI, USA) was used to verify whether the obtained siRNAs do not interact with known human mRNAs. The online application «Oligo Calc» (Northwestern University, USA) was used to check whether the candidate siRNAs form thermodynamically stable elements of secondary structure, such as internal dimmers and pins. The online tool «Palindromic sequences finder» (BioPHP, USA) was used to identify palindromes. Finally, the online application «siRNA Wizard Software» (InvivoGen) was used to convert the sequences of siRNAs to the sequences encoding shRNA.
The DNA concentration was assessed using Qubit fluorometer v1.37 and Qubit dsDNA BR assay kit (Thermo Fisher Scientific, USA) according to the manufacturer’s instructions.
To clone the sequence encoding the desired shRNA into the expression vector pGPV-17019250 (Evrogen, Russia), the vector (1 μg) was exposed to BamH1 and EcoRI restriction endonucleases (1U each) in the commercial buffer R (Thermo Fisher Scientific, USA) for 2h at 37°C. To avoid inhibition of the enzymes, their total volume did not exceed 10% of the reaction mix. Then, the restriction endonucleases were thermoinactivated (20 min; 80°C) and the obtained DNA fragments were separated by electrophoresis in 1% agarose gel.
DNA Electrophoresis was performed in agarose gel. The gel contained 1% agarose, TAE buffer (40 mM TRIS, 20 mM acetic acid, 1 mM EDTA, pH 7.6) and 0.5 μg/mL of ethidium bromide. The samples were prepared in the loading buffer (0.04% bromophenol blue, 10% sucrose and 60 mM EDTA, pH 8.0) to increase their weight and help them to sink into the wells. Moreover, bromophenol blue was used as a reference dye. The electrophoresis was performed at 80 — 125 V (3.2 — 5.0 V/cm between electrodes) until bromophenol blue has migrated 2/3 the length of the gel.
To purify a desired DNA fragment from agarose gel, a piece of the gel containing the DNA was cut with a sharp razor blade and transferred into a water solution using «glass milk». Briefly, a piece of gel was incubated in 6M KJ at 55°C until dissolved (5 — 10 min). The DNA was precipitated on «glass milk» (6,000 rpm, 1 min), washed for three times in «new washing buffer» (100 mM NaCl, 1mM EDTA, 10 mM TRIS-HCl, 50% ethanol, pH 7.5) and eluted in a small volume of bidistilled water (2 × 15 μL).
To anneal two complementary single stranded DNA oligonucleotides, they were dissolved in 10 μL of annealing buffer (10°mM TRIS, 50°mM NaCl, 1°mM EDTA, pH°8.0), heated in a 1 L water bath (95-98°C; 2 min) and gradually cooled down in hot water to room temperature (~30 min).
The mentioned above double stranded oligonucleotide and electrophoretically purified DNA fragment were ligated to each other in an ice bath in the ratio 10:1 using 7.5 U of T4 DNA ligase (Thermo Fisher Scientific) per 1 μg of DNA for 16-18 h, according to the previously optimized protocol [4]. After the ice had melted, the water bath was placed on a lab desk for couple of hours that the temperature could reach 20-22°C.
To obtain competent cells, a portion of E. coli suspension (60 μL) was diluted in 6 mL of LB medium, containing tetracycline (12.5 μg/mL) and incubated on a shaker for 1h (37°С, 160 rpm). The tube was kept on ice for 10 min and then, the bacteria were precipitated by centrifugation (6,000 rpm, 5 min, 4°С). After discarding the supernatant, the pellet was gently resuspended in 0.1 M CaCl2 (350 μL) and kept on ice for another 40 min. Then, the bacteria were centrifugated again (12,500 rpm, 30 s, 4°С) resuspended in a fresh portion of CaCl2 (150 μL) and split equally between three tubes.
To transform E. coli, an aliquot of ligation mix (10 μL) or plasmid DNA (1 μL) was added to the competent cells (50 μL). The obtained suspension was kept on ice for 30 min. Then, the suspension was placed in a thermoblock (42°C) for 2 min and returned to ice for 5 min. The transformed bacteria were resuspended in LB (1 mL) and incubated at 37°С for 1h without shaking. The obtained E. coli suspensions were seeded on LB-agar solid medium containing ampicillin (100 μg/mL) and kept overnight at 37°C.
The plasmid DNA was purified using Plasmid Miniprep kit («Evrogen», Russia) according to the instructions provided by the manufacturer. To verify whether the purified plasmid contained the sites susceptible to the restriction endonucleases BamH1 and EcoRI, the plasmid DNA was treated with the named enzymes and the obtained DNA fragments were separated electrophoretically as described above. To confirm that the plasmids contained the cloned sequence, aliquots of plasmid DNA (10 ng) were amplified with the specific primers EXT-F: 5′-ACG TGA AAT GTC TTT GGA TTT GGG-3′ and EXT-R 5′-CAG AGA GAC CCA GTA GAA GCA-3′. After DNA amplification was completed, the PCR products were separated electrophoretically in 2% agarose gel. To verify whether the desired DNA sequence was free of mutations, one of the DNA samples was sequenced in a local «Evrogen» service center.
Results
To select the sequence that would imitate the binding motif in the molecule of scrambled shRNA we used the sequence 5′-CAG TTT CCA TTC ATC TTC CAA-3′. This sequence is the binding motif of shRNA directed to GB that we previously cloned into the vector pGPV-17019250 [4]. Using the online services that belong to GenScript and InvivoGen, we obtained two candidate sequences (Table 1) and compared their properties. First, we analyzed their composition, and calculated how many individual nucleotides of each type the named sequences contained. We also verified whether individual nucleotides in these sequences were repeated for three and more times in a roll. Second, we verified whether they could produce stable elements of the secondary structure, known as «internal dimers» and «pins» using «Oligo Calc» online tool (Northwestern University). Third, we explored the named sequences with «Blastn» suite (NCBI) to be sure that they did not have a significant sequence homology with known human mRNAs. The obtained results suggested us that both oligonucleotides represented in Table 1 could be used as false binding motifs in the molecule of scrambled shRNA.
Table 1. Comparative analysis of cDNA fragments proposed for the design of scrambled GB shRNA
| Source cDNA | InvivoGen (A) | GenScript (B) | |
| Sequence | CAGTTTCCA TTCATCTTC CAA |
GCTATTCAC ATCCCCTTT ATA |
ACTCCATAT ACTTCGTCT TAC |
| Length, bp | 21 | 21 | 21 |
| Nucleotide composition | A = 5; T = 8; G = 1; C = 7 |
A = 5; T = 8; G = 1; C = 7 |
A = 5; T = 8; G = 1; C = 7 |
| Potential hairpin formation | no | no | no |
| Complementarity | no | no | no |
| Self-annealing sites (2 and more mismatches) | no | no | no |
| Purines (G+C), % | 38.1 | 38.1 | 38.1 |
| Number of repeats in consecutive nucleotides | 3 | 4 | 2 |
| The highest homology with the known human mRNAs, % | 100 (MMP9) | 57 (GPX5) | 62 (DCAF16) |
| Palindromes, >4 bp | No | No | no |
bp – base pair; The official gene symbols given in parentheses indicate on the genes that exhibit highest homology with sequences «A» and «B».
Since both tested cDNA fragments satisfied the criteria for siRNA, we have randomly chosen sequence B and used it to design shRNA. The sequence encoding scrambled shRNA was a double stranded DNA oligonucleotide (Figure 1). The upper (parallel) strand of the DNA was composed of several parts, namely half binding sites of the restriction endonucleases EcoRI and BamH1, mRNA binding (sense) motif, the complementary motif (antisense) and the middle loop that connected «sense» and «antisense» motives to each other. In turn, the lower (antiparallel) DNA strand was complementary to the upper one, except 5′ and 3′ termini because the named endonucleases generate cohesive ends.
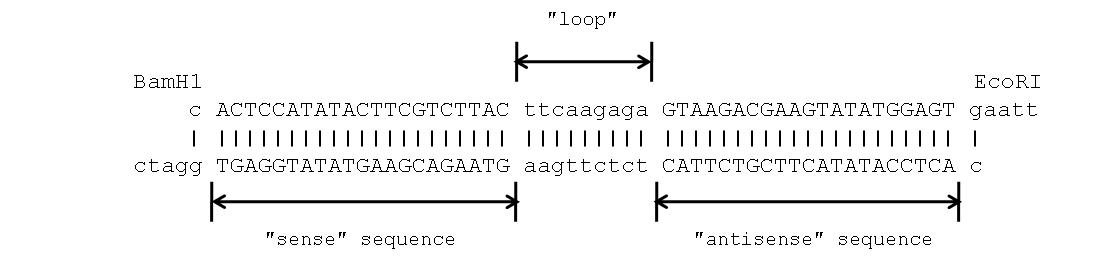
Figure 1. The sequence of double stranded oligonucleotide encoding scrambled GB shRNA.
To clone the mentioned oligonucleotide (Figure 1) into the vector pGPV-17019250 (7,911 bp), the vector was digested with the restriction endonucleases BamH1 and EcoRI. The obtained DNA fragments (7,852 and 59 bp) were separated by electrophoresis in 1% agarose gel (Figure 2) and the larger DNA fragment was purified from the gel. Then, the oligonucleotide and the DNA fragment were ligated and the obtained plasmid was propagated in E. coli.
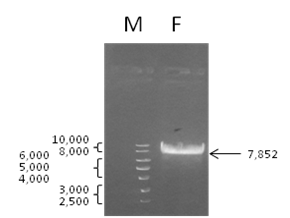
Figure 2. Separation of DNA fragments obtained by digestion of the vector pGPV-17019250 with restriction endonucleases BamH1 and EcoRI. M — 1 kB DNA ladder; F – DNA fragment (7,852 b.p.) designated for ligation and cloning.
The obtained results demonstrated that the procedure that we used to clone the oligonucleotide into the vector was performed with high efficiency. Particularly, we generated several hundred ampicillin-resistant bacterial clones (Figure 3).
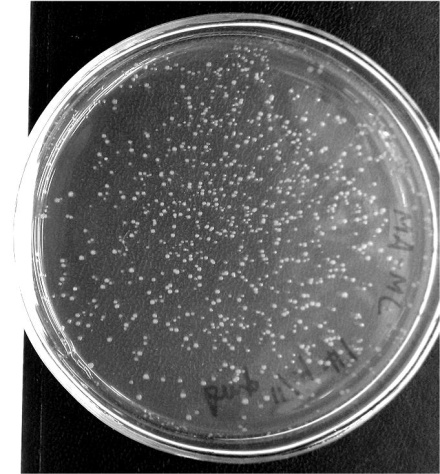
Figure 3. Ampicillin-resistant E. coli clones obtained by transformation of the bacteria with products of the ligase reaction.
To validate the results of cloning, we examined the plasmid DNA isolated from 3 randomly selected clones of transformed bacteria (Figure 4). First, we confirmed that all three DNA samples contained the binding sites of restriction endonucleases BamH1 and EcoRI. We found that both sites were functional and the manipulations that we performed with the vector did not affect the junction regions. Second, we amplified the plasmid DNA by PCR with the specific primers. The DNA amplification revealed the presence of 150 bp PCR-product in the probes. The length of the mentioned PCR-product (Figure 5) was close to the predicted length of the sequence that included the binding sites of the primers and the cloned oligonucleotide (154 bp). Third, sequencing of the plasmid DNA (Figure 6) confirmed that the purified plasmid DNA contained the sequence of the annealed double stranded oligonucleotide.
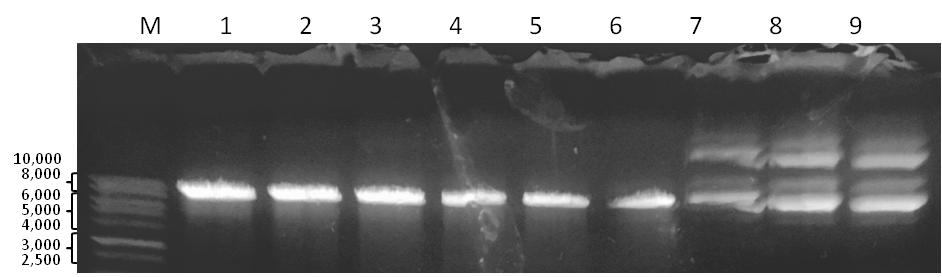
Figure 4. Analysis of the purified plasmid DNA with restriction endonucleases BamH1 and EcoRI. М — 1 kB DNA ladder; 1-3 – plasmid DNA digested with BamH1; 4-6 – plasmid DNA digested with EcoRI; 7-9 – undigested plasmid DNA.
Discussion
A group of non-coding RNAs known as siRNAs interferes with protein biosynthesis in the cell. Interacting with mRNA that encodes a protein of interest, siRNAs cause its degradation and prevent the translation of mRNA into polypeptide by ribosomes. In cell biology, the synthetic siRNAs, known as shRNAs, are often used to silence disease-associated genes. For instance, shRNA specific to MMP9 can be used to reduce accumulation of gelatinase B in the culture medium – in vitro or ECM – in vivo.
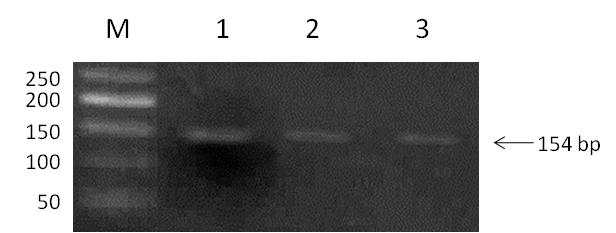
Figure 5. PCR-amplification of plasmid DNA isolated from transformed E. coli. M – 100 b.p. DNA ladder; 1-3 – samples of plasmid DNA from selected clones with amplified PCR-product (154 b.p.).
Although gene silencing with shRNAs is a part of routine lab practice, their overexpression in cells may cause a problem similar to drug overdose. In fact, there is always a certain sequence homology between shRNA of choice and non-target mRNAs (Table 1). Respectively, than more shRNA the cell produces than higher probability of its interaction with non-target mRNAs exists. This kind of non-specific interactions is known as «off target effects».
In turn, an obvious way to distinguish «off target effects», which are caused by interactions of specific shRNA with non-specific mRNAs from specific effects, which are caused by interaction of the same shRNA with mRNAs encoding the target protein would be to compare two kinds of cells expressing specific and scrambled shRNA, respectively. For these proposes, the scrambled shRNA should have the nucleotide composition identical to the specific shRNA and should not interact with any known mRNA. Respectively, there is a need in a vector that would express scrambled shRNA.
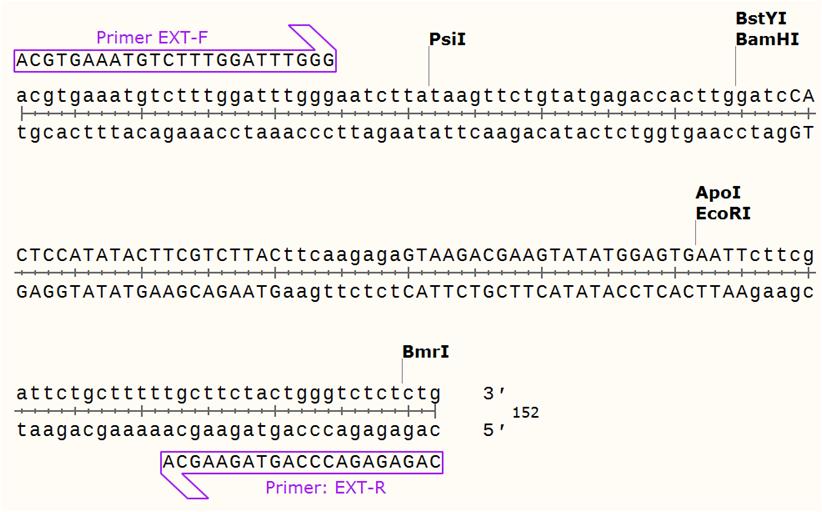
Figure 6. Sequences of plasmid DNA isolated from the transformed E. coli. The cloned oligonucleotide is located between the binding sites of restriction endonucleases BamH1 and EcoRI.
To obtain the expression vector that encodes scrambled shRNA, we chose the sequence 5′-CAG TTT CCA TTC ATC TTC CAA-3′, which is a part of exon 6 in the gene of human gelatinase B. According to the analysis that we previously performed, this cDNA fragment is susceptible to GB specific shRNAs [4]. We already used this sequence to design a GB specific shRNA and obtain the expression vector pGPV-17019250-GB encoding the named shRNA. In this paper, we used the same sequence to design scrambled shRNA and clone it into the vector pGPV-17019250. Respectively, we obtained the expression vector pGPV-17019250-KGB encoding the designed scrambled shRNA.
First, we obtained two sequences of candidate cDNAs (Table 1) using the common online tools. Second, we explored their properties. Particularly, we compared their nucleotide compositions and analyzed their secondary structures (Table 1). Third, we selected one of the qualified cDNAs (Table 1, sequence B), cloned it into the expression vector pGPV-17019250 and obtained a new vector, namely pGPV-17019250-KGB. Using the method of automatic DNA sequencing, we also confirmed that pGPV-17019250-KGB contained the sequence encoding the chosen scrambled shRNA (Figure 5).
To clone the selected scrambled shRNA (Figure 1), we exposed the vector pGPV-17019250 to the restriction endonucleases EcoRI and BamH1. Particularly, we chose these two enzymes because we wanted to preserve the integrity of 5′-LTR – sequence and H1 promoter, which are important for functioning of the vector. Particularly, the intact H1 promoter is necessary to induce the shRNA expression in human cells and 5′-LTR sequence is needed to integrate the lentiviral genome encoded by pGPV-17019250 into the genome of the infected cell.
We would also acknowledge a high efficiency of the method that we used to transform E. coli. This is one of several known protocols commonly used to introduce plasmid DNA into bacteria [7]. Since these protocols use different chemical agents (e.g. CaCl2, DMSO and PEG) to produce competent cells, different conditions for their transformation and different procedures to recover the transformed cells, their performance varies from strain to strain.
In this paper, we used the calcium chloride method to transform Xl1 blue E. coli strain [9]. This strain is one of the six most frequently used E. coli strains to propagate plasmids with antibiotic-resistant factors in bacteria [7]. This strain was originated from K12 strain that, in turn, was isolated from a diphtheria patient [1]. Due to its high transfection efficiency (~1 × 108 cfu/µg), the Xl1 blue strain is recommended by the manufacturers for routine cloning applications [9] including one that we have performed. Although we are satisfied with transformation efficiency that we have achieved in our experiments (Figure 3), according to the others [7] the results still could be improved if we replaced calcium chloride by DMSO and polyethylene glycol and used Hanahan’s protocol [3] to transform the bacteria.
To confirm that the shRNA encoding sequence is properly cloned into the expression vector pGPV-17019250, we examined 3 randomly selected clones of transformed E. coli. Particularly, we wanted to see whether the plasmids that we purified from bacteria contained the binding sites of restriction endonucleases BamH1 and EcoRI. In the other words, we wanted to be sure that the sites that we used to clone the shRNA encoding sequence into pGPV-17019250 preserved their integrity. It was necessary to avoid mutations that the vector could gain in the process of cloning. We generated two sets of DNA samples (three samples per a set) and exposed them to either BamH1 or EcoRI. Then, we separated the obtained DNA fragments in agarose gel as described above. Our results suggested us that all three mentioned samples of plasmid DNA were sensitive to both restriction endonucleases (Figure 4). Moreover, we found that the length of the digested DNA was about the same to one that we anticipated to see (~7,800 bp). Thus, all three tested clones could be used for further testing.
The commercial T4 DNA ligases designated for molecular cloning are genetically modified to increase their accuracy and reduce the mutation rate. Unfortunately, even accurate enzymes make mistakes and produce mutations. For instance, T4 DNA ligases have the ability to delete modified nucleotides if they are occasionally present in the DNA [2]. In this respect, if a synthetic oligonucleotide is used, their presence is more likely due to possible technical problems with DNA synthesis. Moreover, the error rate depends on the concentration of ATP in the reaction mix. Particularly, than ATP concentration is lower than the mutation rate is higher and the nucleotides that directly interact with the enzyme are at a higher risk. For instance, ATP partially degrades if the ATP stock solution is thawed and frozen for few times [5]. Unfortunately, it often happens in the lab where many people have unlimited access to the ATP stock.
Since the BamH1 and EcoRI binding sites that we verified were intact, we wanted to be sure that the length of the DNA that we inserted into the vector was correct. Respectively, we amplified the plasmid DNA by PCR. For PCR amplification, we used the specific primers that flanked the insert in the vector. The results of PCR amplification demonstrated that the length of the obtained PCR product (~150 bp) and the estimated length of DNA insert (154 bp) were about the same (Figure 1 and 5).
To confirm that the cloned DNA encodes the desired shRNA, we sequenced one of the tested DNA samples with the vector-specific primers mentioned above. Importantly, we sequenced both DNA strands using one primer per probe. Respectively, each obtained DNA sequence contained a fragment identical to the cloned oligonucleotide. Moreover, both DNA sequences overlapped to each other. The latter let us explore the junction regions of the cloned oligonucleotide and the vector. Based on the sequencing results we found that the tested plasmid encoded the desired shRNA.
In conclusion, we would like to acknowledge that we obtained the vector pGPV-17019250-KGB encoding scrambled shRNA. This vector could be used as a negative control in silencing human gelatinase B in vivo and in vitro. We also obtained clones of the transformed E. coli that carried the named vector. These clones can be used to propagate the vector pGPV-17019250-KGB in bacteria.
References
1. Bachmann B.J. Pedigrees of some mutant strains of Escherichia coli K-12 / B.J. Bachmann // Bacteriol. Rev. –1972. – V. 36. – № 4. –P. 525-557.
2. Bogenhagen D.F. The action of DNA ligase at abasic sites in DNA / D.F. Bogenhagen and K.G. Pinz// J. Biol Chem.– 1998. – V. 273. – № 14. –P. 7888-7893.
3. Chan W.T. A comparison and optimization of methods and factors affecting the transformation of Escherichia coli / W.T. Chan, C.S. Verma, D.P. Lane and S.K. Gan // Biosci Rep. –2013. – V. 33. – № 6. – pii: e00086.
4. Green M.R. and Sambrook J. The Hanahan Method for Preparation and Transformation of Competent Escherichia coli: High-Efficiency Transformation // Cold Spring Harb. Protoc. –2018. – № 3. –pdb.prot101188.
5. Green M.R. and Sambrook J. Molecular Cloning: A Laboratory Manual: 4th ed.: Cold Spring Harbor: Cold Spring Harbor Laboratory Press, 2012. – P. 235-239.
6. Mezentsev, A. Matrix metalloproteinases and their role in psoriasis / A. Mezentsev, A. Nikolaev, and S. Bruskin //Gene. – 2014. –V. 540. – № 1. – P. 1-10.
7. Mogulevtseva J.A. Cloning a sequence of small hairpin RNA directed to human gelatinase B into the expression vector pGPV-17019250 / J. Mogulevtseva and A.V. Mezentsev // Wschodnioeuropejskie Czasopismo Naukowe. –2019. – № 44. – Pt. 1. –P. 32-38.
8. Starodubtseva N.L., Sobolev V.V., Soboleva A.G. et al. // Expression of genes for metalloproteinases (MMP-1, MMP-2, MMP-9, and MMP-12) associated with psoriasis Russ. J. Genet. – 2011. –V. 47. – №. 9. – P. 1117–1123.
9. XL1-Blue Competent Cells, manual [Electronic resource]. URL: https://www.chem-agilent.com/pdf/strata/200249.pdf (Accessed at 21.05.2019).