

Постановка задачи
В лаборатории молекулярного моделирования и спектроскопии ГЕОХИ поставлена задача прямого моделирования химической кинетики средствами вычислительной техники на основе физических представлений и принципов. В данной работе исследуется вопрос – допустимо ли и достижимо ли предсказать скорости протекания реакций в сложной химической системе, опираясь лишь на физические представления об элементарных процессах в системе. То есть, ищутся такие физические и математические средства, которые могут позволить априорную картину химической кинетики без привлечения опытных данных о скоростях отдельных реакций в системе и без составления сложных кинетических уравнений. Предполагается, что на этом пути удастся прояснить все физические факторы, формирующие полную кинетическую картину превращений в химической системе.
В теоретической и индустриальной химии разработан известный подход к прогнозированию сложных процессов в химических реакторах, основанный на составлении разветвленной системы дифференциальных уравнений для скоростей реакций с учетом эмпирических констант. Последнее как раз и составляет проблему, для решения которой в случае реакций второго и более высоких порядков приходится прибегать к громоздким методам хемометрики, причем без гарантии получения необходимой для практики точности решения обратных задач.
В данной работе проверяется возможность прямого следования природной логике микроскопического акта химического превращения, а макроскопические следствия из физических представлений получать статистическими методами.
Теоретические предпосылки
Наши представления о физических факторах, определяющих химическую кинетику, сводятся к следующему.
- Вступающие в реакцию молекулы должны найти друг друга в пространстве, перебирая различные сочетания типов молекул в случайных парных столкновениях.
- В ходе столкновения две подходящие молекулы должны расположиться друг относительно друга удачным для реакции образом. Благоприятны лишь такие расположения молекул в предварительно возникающем комплексе, когда реакция может осуществиться путем минимальных перемещений минимального количества атомов. Тогда возникает реакционный центр во временном комплексе [1, 2].
- Колебательные состояния обеих молекул должны обеспечить высокую вероятность структурной перестройки ради получения желаемого продукта [2, 3].
- Продукты реакции, если это не единственная молекула, должны иметь возможность разойтись в пространстве, чтобы тут же не произошла обратная перестройка структур в исходные, иначе факт элементарного акта реакции не будет заметен в макроскопических измерениях.
Обсудим наличие или отсутствие готовых средств для теоретического анализа и для моделирования этих факторов в компьютерном эксперименте.
- Фактор необходимых парных столкновений
Столкновение молекул-участниц бимолекулярной реакции есть совершенно необходимое условие элементарного акта химического превращения, но это не достаточное условие. Статистическая физика располагает средствами предсказания частоты столкновений молекул в газе любой плотности. Однако это весьма ограниченные средства. Они хороши для случая ансамбля одинаковых молекул, но не годятся для случая большого разнообразия типов молекул в ансамбле. Не говоря уже о необходимости учета форм молекул, чего статфизика вообще никогда не принимала во внимание.
Для анализа факторов химической кинетики такой подход к делу совершенно не годится, поскольку тут важны именно подробности процессов. Строя макроскопическую картину хода реакций в сложной химической системе, мы должны поступать так, как поступает сама Природа, считающая всё на штуки. Важно знать, молекулы каких именно типов столкнулись, с какими энергиями, в каком вращательном и колебательном состоянии. Важно знать, какое время может прожить промежуточный реакционный комплекс, не подвергаясь воздействию налетающих на него сторонних молекул. Важно знать, что ждет молекулу-продукт реакции в течение короткого промежутка времени после окончания перестройки реакционного комплекса, не будет ли она снова притянута к остальным продуктам реакции. В этом случае акт реакции может быть аннулирован актом обратной реакции. А в случае фотохимической реакции важно знать, нет ли вероятности попадания нового кванта света в такую молекулу, что может драматически повлиять на судьбу продукта.
Отметим, что уже пятьдесят лет развивается новая отрасль физической статистики. Это молекулярная динамика. Важнейшая особенность этой статистики состоит в том, что она в процессе слежения за молекулярными процессами не теряет ни одной единицы статистической информации. Она ведет полную статистику для огромного молекулярного ансамбля в течение короткого времени Сегодня рекордное время правильного интегрирования уравнений движения для тысяч молекул с полным учетом всех взаимодействий между ними и с учетом внутренних молекулярных движений – это одна микросекунда. Этого вполне достаточно для накопления всех необходимых сведений о происходящих в ансамбле явлениях. Если же учитывать не все взаимодействия и не все формы взаимодействующих молекул, то время слежения за процессами может быть и более продолжительным.
Мы выбрали молекулярную динамику (МД) как средство слежения за подробностями молекулярных процессов, имеющих отношение к химическим реакциям. К сожалению, мы не нашли готового подходящего варианта молекулярной динамики. ХемОфис в коллекции программ ММ2 содержит готовые программы МД. Но они не позволяют задавать начальные скорости молекул. Приходится создавать нужные программы, опираясь на мировой опыт молекулярной динамики в плане техники составления и решения уравнений движения.
Выяснилось, что целесообразно иметь два связанных варианта программ МД. Программы первого эшелона предназначены для слежения за свободными полетами молекул между столкновениями. Программы второго эшелона предназначены для подробного анализа событий во временном комплексе столкнувшихся молекул. Эти программы получились довольно сложными не только в части структуры их алгоритмов, но и в плане их параметризации.
Выяснилось, что даже для моделей очень плотного газа можно не учитывать силы притяжения между молекулами, включая их затем только в момент непосредственного соприкосновения сфер ван дер Ваальса. А распознав парное столкновение, программа МД должна передать столкнувшуюся пару молекул подпрограмме химического процессора. Последний должен определить, подходящие ли это типы молекул для реакции заданного типа. Если нет, то пара возвращается в ансамбль, обменявшись скоростями по законам столкновения абсолютно упругих шаров. Если да, то химический процессор проверяет достаточность условий для структурного превращения и заменяет пару продуктами реакции.
Первые программы строго сохраняют полную энергию системы в течение времени в несколько наносекунд. Вторые программы сохраняют энергию и момент импульса пары молекул в случае длительного движения пары молекул в общей потенциальной яме. При выполнении структурной перестройки с изменением числа молекул (реакции слияния или разложения) энергия участников реакции меняется. Тогда программа первого эшелона восстанавливает температуру новых молекул до средней, имитируя действие термостата.
В программах первого эшелона в ансамбле сохраняется заданное вначале распределение Больцмана по энергиям всех молекул, в том числе и появившихся в результате реакций. Более того, был поставлен эксперимент, когда всем молекулам придавали одну и ту же энергию поступательного движения, соответствующую заданной температуре. Через короткое время обнаруживалось, что в результате случайных столкновений гистограммы распределения молекул всех типов по энергиям становятся очень похожими на ожидаемые по закону Больцмана.
Сконструированные программы МД вполне правильно воспроизводят привычную картину хаотического теплового движения молекул в газе любой плотности. Эти программы мы принимаем за теоретическую базу, обеспечивающую адекватную имитацию действия первого фактора из приведенного выше перечня.
- Фактор благоприятного взаимоположения молекул в реакционном комплексе
Этот фактор совершенно невозможно прояснить теоретическими построениями в случае молекул сложной пространственной формы. Поэтому мы пошли по пути прямого моделирования механического поведения пары молекул в процессе их столкновения.
Приведем простой пример. Он относится к модели близко расположенных молекул водорода. Расположим их так, чтобы они налетали друг на друга поступательно, оставаясь параллельными друг другу. Пусть одна молекула сдвинута вдоль своей оси на длину самой молекулы. Тогда молекулы при сближении должны удариться только крайними атомами.
Заданы начальные скорости движения молекул навстречу друг другу. Эти скорости, 2 см/с, совершенно ничтожны по сравнению со скоростями молекул в тепловом движении. Введем обобщенную координату d как изменение расстояния между центрами масс молекул по сравнению с равновесным расстоянием d0 = 1 Å. На рисунке 1 показана картина классических ангармонических колебаний в этой системе.
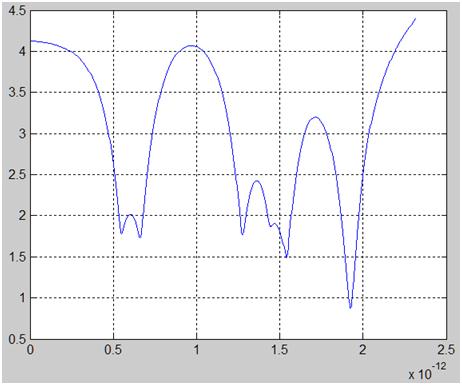

Рисунок 1. По оси абсцисс отложено время жизни комплекса из двух молекул водорода, с. По оси ординат – расстояние между их центрами масс, Å.
Далее, молекулам задавали начальные скорости, соответствующие средней энергии теплового движения. Например, поступательная скорость принималась равной 1.8 км/с. При такой начальной энергии столкновения время жизни комплекса получается сравнительно небольшим. Однако в системе наблюдаются неоднократные соударения атомов, что может обеспечить прохождение реакции изотопного обмена.
Написана программа визуализации поведения молекул в столкновениях. Эта программа позволила внимательно рассмотреть весь процесс для более сложных молекул и сделать некоторые выводы, имеющие общий характер.
В случае не лобового столкновения молекул в системе имеется и сохраняется вращение. При этом скорости вращения молекул изменяются по величине и по знаку, оставляя неизменной общую механическую энергию молекул. В результате характер движения становится нерегулярным. Но самое главное, молекулы при многократных встречах соударяются различными парами атомов, либо несколькими парами атомов. При этом нередко возникают различные весьма тесные взаимные расположения молекул. Молекулы не только крутятся между соударениями, но и перекатываются друг по другу, создавая возможности нащупать нужную для реакции конфигурацию промежуточного комплекса. При этом не очень важна начальная энергия столкновения молекул. При колебаниях в яме ван дер Ваальса молекулы разгоняются и раскручиваются силами их взаимодействия до скоростей, сравнимых со скоростями теплового движения.
Таким образом, сформирована теоретическая база для прояснения роли первых двух физических факторов в формировании химической кинетики.
- Фактор колебательных состояний молекул
Анализ этого фактора облегчается тем, что скорости колебательных движений атомов в молекулах значительно выше скоростей молекул, испытывающих соударения. Поэтому можно забыть о поступательных и вращательных движениях молекул как целого, расположить их так, чтобы образовался реакционный центр (как их располагает иногда сама механика столкновений), а затем подробно проанализировать роль колебаний в образовавшейся системе.
Последовательная квантовая теория элементарного акта химического превращения изложена в [2, 3]. Заинтересованный читатель может с ней познакомиться самостоятельно. Важно, что при анализе двух первых факторов выяснилось – в не слишком плотных средах столкновение подходящей пары молекул обязательно приведёт к нужному для реакции взаимному расположению молекул. Поэтому вероятность нужного для реакции столкновения определяется только первым фактором, а численная величина вероятности этого продуктивного столкновения выясняется методами МД. Для определения полной вероятности прохождения одного элементарного акта реакции достаточно дополнительно вычислить вероятность превращения методами [2, 3].
Приведём результаты полного численного расчёта кинетики накопления промежуточных комплексов из двух молекул пропилена, приводящих к реакции образования четырёхчленных циклов как инициаторов олигомеризации пропилена [2, с. 378].
В кубическом объеме с идеально отражающими стенками случайно разместили 720 молекул пропилена, придали им случайные скорости по Больцману при температуре 400 К и поручили их программе молекулярной динамики. На рисунке 2 показано, как со временем, выраженном числом всех случайных соударений молекул, уменьшается популяция мономеров пропилена и растёт популяция комплексов ван дер Ваальса, дающих нужные для олигомеризации четырехчленные циклы в димерах пропилена.


Рисунок 2. Кинетика образования комплексов пропилена, ведущих к олигомеризации пропилена (зелёная кривая).
Время на рисунке 2 отсчитывается числом всех соударений молекул в системе. Это позволяет непосредственно подсчитать вероятность акта реакции на одно столкновение и изменение числа актов на одно столкновение в зависимости от популяций компонентов системы.
Из рисунка видно, что получен вполне ожидаемый результат, причем он получен без составления дифференциальных уравнений и заимствования констант реакции из эксперимента. При увеличении объёма системы и сохранении начальной плотности газа кинетические кривые становятся всё более гладкими. Это позволяет перевести результаты на язык дифференциальных уравнений и предсказать скорость реакции чисто теоретическим путём, не прибегая к химическому опыту. Важно, что описанная техника никак не меняется в случае, когда в системе происходят химические реакции высоких порядков.
Второй пример, показанный на рисунке 3, – это кинетика изотопного обмена в сверхплотном газе водорода. Задан кубический объем, состоящий из 8 тысяч кубических ячеек, каждая с ребром в 2 Å. Клеточный автомат в одну ячейку может поместить лишь одну молекулу водорода. Программа случайным образом размещает по ячейкам 400 молекул H2 и 400 молекул HD. Ожидается появление молекул D2 в результате изотопных обменов в ходе столкновений молекул. Популяция молекул D2 вначале равна 0. Температура T=300 К. Программа распределяет молекулы по энергиям в соответствии с законом Больцмана и с заданной температурой. При встрече двух молекул HD программа с вероятностью 0.25 заменяет эти две молекулы молекулами H2 и D2 независимо от скоростей молекул. При встрече молекул H2 и D2 программа заменяет их двумя молекулами HD. Скорости и направления движения новых молекул определяются законами сохранения энергии и импульса.
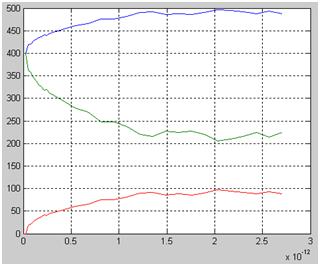

Рисунок 3. Кинетика изотопного обмена в смеси водорода и тяжелого водорода. Популяции, сверху вниз: H2, HD и D2. Время в с.
Заключение
В работе описаны предварительные результаты компьютерных имитационных экспериментов по прогнозированию физическими методами химической кинетики. Подробности построения алгоритмов программ молекулярной динамики, их параметризации и анимационные картины столкновений молекул приведены в блоге «Физические факторы химической кинетики» на сайте ГЕОХИ [4]. Там же приведены все подробности физического обоснования разработанных алгоритмов прогнозирования химической кинетики. Намечены дальнейшие стадии исследования физических аспектов химических превращений и запланированы более сложные вычислительные эксперименты.
Список литературы:
- Грибов Л.А. От теории спектров к теории химических превращений. М.: УРСС, 2001. — 365 с.
- Грибов Л.А., Баранов В.И. Теория и методы расчета молекулярных процессов. Спектры, химические превращения и молекулярная логика. М.: URSS, 2006. — 476 с.
- Грибов Л.А., Баранов В.И., Дементьев В.А. К вопросу о теории процессов в реакционных центрах многоатомных молекул. Известия Академии наук. Серия химическая. 2006, №8, — 1267-1273.
- Физические факторы химической кинетики. Блог сайта ГЕОХИ. type=»book» name=»ФИЗИЧЕСКИЕ АСПЕКТЫ ХИМИЧЕСКОЙ КИНЕТИКИ» author=»Дементьев Василий Александрович» publisher=»БАСАРАНОВИЧ ЕКАТЕРИНА» pubdate=»2017-04-22″ edition=»ЕВРАЗИЙСКИЙ СОЮЗ УЧЕНЫХ_ 28.03.2015_03(12)» ebook=»yes» ]